Thermodynamic Stability and Anion Ordering of Perovskite Oxynitrides
19 Jul 2023·
 ,
,
,
,
·
1 min read
,
,
,
,
·
1 min read
Samuel D. Young
Jiadong Chen
Wenhao Sun
Bryan R. Goldsmith
Ghanshyam Pilania
Abstract
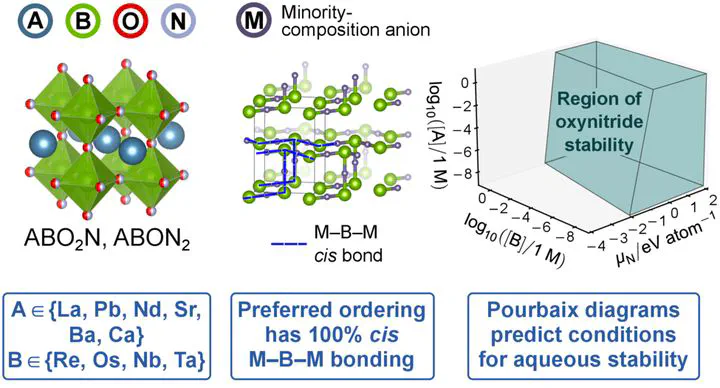
Perovskite oxynitrides (PONs) are a promising class of materials for applications ranging from catalysis to photovoltaics. However, the vast space of single PON materials (ABO3-xNx) has yet to be fully explored. Additionally, the community needs guidelines that relate PON chemistry and anion ordering to stability to better understand how to design PON materials that resist corrosion and decomposition under operating conditions. Screening this materials space requires identifying candidate PON materials that will be stable under operating conditions, which in turn requires methods to evaluate each material’s stability. Here we predict the stability of single PON materials using a four-step approach based on density functional theory modeling: (i) enumerate viable cation pairs, (ii) select an energetically favorable prototypical anion ordering, (iii) compute each PON’s energy above the thermodynamic convex hull, and (iv) generate computational Pourbaix diagrams to determine allowable ranges of electrochemical operating conditions. A critical part of our approach is determining a prototypical stable anion ordering for both ABO2N and ABON2 stoichiometries across a variety of A- and B-site cations. We demonstrate a stable anion ordering containing a high degree of cis ordering between B cations and minority-composition anions. We predict 85 stable and 109 metastable PON compounds, with A = {La, Pb, Nd, Sr, Ba, Ca} and B = {Re, Os, Nb, Ta} forming cation pairs that lead to stable PONs less than 10 meV/atom above the thermodynamic convex hull. Computational Pourbaix diagrams for two stable candidates, CaReO2N and LaTaON2, suggest that not all compounds with zero energy above the thermodynamic convex hull can be easily synthesized.
Type
Publication
Chemistry of Materials